10 Years of Modality Solutions: What We’ve Learned, Part 2
In our previous post, we looked at some of the ways the biopharmaceutical industry...
read Details

First published on September 16, 2015 on Cold Chain IQ – as a contributor, Hutchinson’s column, Global Cold Chain Connections, falls in the Supply Chain & Security category. It can also be viewed at http://www.coldchainiq.com/regulatory-resources/columns/a-process-validation-guide-for-cold-chain-logistic
Process validation for cold chain logistics (packaging, storage, and distribution) is required part of the Common Technical Document (CTD) for any Biologics License Application (BLA) for monoclonal antibodies. Any review of the submitted dossier and subsequent pre-approval inspection on site will most likely review the following areas:
The strategies focused on the packaging qualification and do not consider validating the process of drug product transport and the potential impact on the potency, efficacy, or purity of the drug product as compared to the specification will be challenged by regulatory agencies around the world. The best practices identified and current expectation from regulatory agencies globally is drug product quality will be confirmed after transport.
The confirmation of drug product quality can be confirmed by a variety of methods. Either simulated or actual shipments of drug product can be used to confirm quality. However, in both cases the confirming test must be justified. The justification can be either a comparison of a proposed testing standard (e.g. ASTM-4169) to actual shipping lanes or the choice of a representative lane that provides a suitable challenge based on the expected channels of distribution. A simulation strategy is ideally shared with the regulatory agencies at the “End of 2A” meeting; otherwise actual shipments within longest lanes may be the expectation. However, any proposed testing methodology that is justifiable and sufficient for the BLA and can be used as supporting documentation for PAI.
Tying your distribution studies back to your excellent stability studies results provide excellent justification for either:
Recombinant produced monoclonal antibody (mAb) products usually contain size variants (e.g., aggregates, fragments) that are generated during manufacture, storage and distribution. Because aggregates and fragments may potentially affect immunogenicity and potency, their levels are typically monitored during lot release, stability, and characterization. These size variants can be exaggerated during transport at elevated temperatures. Any proposed engineering study or follow on process validation should include enough data points to confirm trending on this key stability indicating method.
Thermal packaging qualification testing for the BLA submission should demonstrate that the selected thermal shipping system provides adequate thermal protection for the entire manufacturing process including shipments to the distribution centers. The FDA regulatory scope starts at point of manufacturer and ends at the point of distribution for the BLA. Representative shipping points that can be justified can be used. The recommended critical-to-quality attributes are:
Qualification of the shipping system to maintain thermal and payload integrity during the distribution process requires a several components:

The principles of qualification for the transport of temperature-sensitive medicinal products closely follow established guidelines and regulations for qualifying the manufacture of these same products as outlined in the PDA Technical Report #39: Guidance for Temperature-Controlled Pharmaceuticals. These include:
Medicinal products are transported in a commercial environment as opposed to a controlled laboratory environment. Factors such as unforeseen transport events and the weather affect the actual conditions a specific shipment may encounter. These factors should be considered when designing test protocols and in understanding “anticipated extreme” challenges. Utilizing this type of information (i.e. typical transportation extremes) to support widening shipping temperature range versus label claim storage conditions is beneficial when supported by product stability data. The last two elements (ongoing monitoring and/or periodic evaluation and change control) are not addressing the shipping study plan, but should be addressed procedurally as part of an integrated cold chain management quality system. A comparison of your current testing approach to the recommended approach the Technical Report #39 is a good place to start any initial assessment. You can use a checklist to determine you preparedness. Table 1 below shows the elements of the shipping qualification and compares them to the proposed test plan.
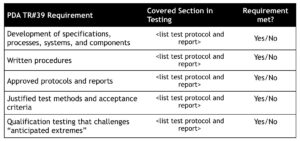
The two requirements (justified test methods and “anticipated extremes” challenge) are symptoms of the same root cause. Your testing methodology must be justified as the proper testing approach for the distribution environment. Since a specific testing approach (e.g. ASTM D 4169) may be a generally accepted test procedure, the lack of justification of the test method and the associated acceptance criteria is low risk. Separating the engineering study from the OQ in any testing plan is recommended. DO NOT START QUALIFICATION until you have confirmed results in with engineering studies. These studies can be already executed as part of characterization or may have to be designed from scratch if the previously executed studies
The Validation Master Plan for Product Distribution (VMP) describes the policies and strategies of the qualification program for product shipment qualification and defines the requirements for the storage and transport of Drug Substance (DS), Bulk Drug Product (BDP), and finished Drug Product (DP) manufactured at company-operated sites or approved contracted manufacturing sites. While there is no explicit statutory requirement by the FDA for a VMP, the 2011 Guidance for Industry for Process Validation implicitly recommends one. A VMP is an expectation in the EU jurisdictions.
The scope of the VMP includes the storage and transport of DS, BDP, and DP originating from facilities, contract manufacturing sites, and/or contract storage facilities to contract manufacturing facilities, commercial packaging facilities, distribution centers, commercial customers (pharmacies), and clinical packaging facilities. Shipping qualification activities include, but are not limited to, the qualification of packaging configurations, temperature profiles, shipping carriers and the use of temperature monitoring devices. In general the VMP includes the storage and transportation activities for each transport segment. Good practice in a VMP is to include a figure representing the distribution process.
The validation master plan overall must justify how DS, BDP, and DP are shipped and stored in a manner that prevents damage and minimizes exposure to temperatures outside of their recommended storage conditions that could potentially impact the safety, quality and effectiveness of the drug product. A validation master plan provides guidelines, requirements and scope for all existing and future shipping configurations, carriers and distributors managed by the manufacturer. The VMP reflects the external and internal standards and guidance documents to comply with Good Distribution Practices (GDP) in addition to other regulations and guidance for the distribution points in the supply chain. Overall, the VMP describes the documentation and qualification activities for all shipping configurations including DS, BDP, and DP. The principles of process validation (including design, operational and performance qualifications) are applied to GDPs such that the portions of the supply chain are qualified (excluding unforeseen transport events and the impact of extreme weather conditions).

In our previous post, we looked at some of the ways the biopharmaceutical industry...
read Details
Recently, we worked on a project for an advanced CAR T-cell therapy developed by...
read Details
First published on January 5, 2016 on Cold Chain IQ – as a contributor,...
read Details